 |
Peter Hugo Nelson, Ph.D.
B.Sc. Victoria University of Wellington (1984)M.Sc. Victoria University of Wellington (1990)
Ph.D. M.I.T. (1998)
|
Peter Hugo Nelson, Ph.D.B.Sc. Victoria University of Wellington (1984)M.Sc. Victoria University of Wellington (1990) Ph.D. M.I.T. (1998) |
Surfactants or "surface-active-agents" are a class of
 molecules that we all use everyday to clean household items and ourselves. Soap and detergent molecules are surface active because they have a "head" region (gray beads in figure) that likes water (is hydrophilic) and a oily "tail" region (red beads) that hates water (is hydrophobic). To accommodate these two regions, the surfactant molecules tend to sit at interfaces between water and another phase (such as oil or air). In dishwashing, the surfactant molecules migrate to the surface of oily residues, surrounding them with a hydrophilic layer of the exposed head groups, thereby making the oil soluble in water. The surfactant also reduces the surface tension of the oily deposits allowing them to breakup more easily. Once the oil/water air/water interfaces are saturated with surfactants the excess self-assembles into spherical and cylindrical surfactant aggregates known as micelles (see figure). Industrial applications of surfactants range from enhanced oil recovery and cleanup of ground water contaminated with pollutants, to stabilizing cosmetics and food stuffs. Self-assembled polymer and surfactant systems are also the molecular basis of soft tissue in living things. For example the cell membrane is a self-assembled bilayer of surfactant molecules, and proteins are polymers that are self-assembled into structures that have biological utility.
molecules that we all use everyday to clean household items and ourselves. Soap and detergent molecules are surface active because they have a "head" region (gray beads in figure) that likes water (is hydrophilic) and a oily "tail" region (red beads) that hates water (is hydrophobic). To accommodate these two regions, the surfactant molecules tend to sit at interfaces between water and another phase (such as oil or air). In dishwashing, the surfactant molecules migrate to the surface of oily residues, surrounding them with a hydrophilic layer of the exposed head groups, thereby making the oil soluble in water. The surfactant also reduces the surface tension of the oily deposits allowing them to breakup more easily. Once the oil/water air/water interfaces are saturated with surfactants the excess self-assembles into spherical and cylindrical surfactant aggregates known as micelles (see figure). Industrial applications of surfactants range from enhanced oil recovery and cleanup of ground water contaminated with pollutants, to stabilizing cosmetics and food stuffs. Self-assembled polymer and surfactant systems are also the molecular basis of soft tissue in living things. For example the cell membrane is a self-assembled bilayer of surfactant molecules, and proteins are polymers that are self-assembled into structures that have biological utility.
A key feature of this research has been developing simulation techniques for systems that can not be modeled using traditional simulation techniques such as molecular dynamics (MD). In principle, MD simulations could investigate any classical problem at a molecular-level, but the time scale of MD is determined by real-world molecular motions. These occur very rapidly at room temperature, so that MD is limited to processes that occur on the order of femto- to nano-seconds. In contrast, many systems of technological interest and most biological systems have intrinsic time scales on the order of milliseconds to hundreds of seconds or longer, and are thus not amenable to direct MD simulation using current computer technology, or with technology expected within the next 50 years.
The first kinetic Monte Carlo (kMC) simulations of adsorption and diffusion in zeolites suffered from systematic errors. This type of simulation was placed on a solid theoretical foundation by the author and co-workers in 1989. 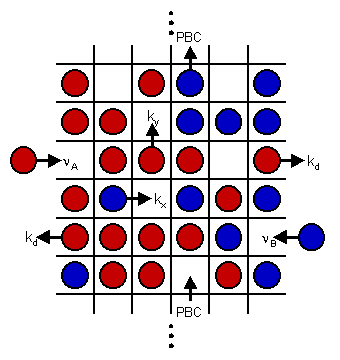 Since then, the technique has gained wide-spread use in the zeolite literature where a wide variety of zeolite problems have been investigated ranging from the basic problem of single component diffusion through multi-component systems, reactive catalytic systems and catalyst deactivation. Recently we have returned to the issue of tracer diffusion in model zeolites (see figure). A key feature of this problem is that Fick's law of diffusion becomes a matrix equation with two eigenmodes, one corresponding to single component transport diffusion (co-diffusion) and the other to self-diffusion (counter-diffusion).
A steady-state method, tracer counter-permeation, (TCP), has been developed for investigating the counter-diffusion eigenmode of diffusion. The advantage of TCP over all other methods is that the counter-diffusion eigenmode (corresponding to self-diffusion) can be investigated directly using Fick's law under steady state conditions. Recently, we have investigated TCP in anisotropic zeolites using kMC. These studies have shown that TCP in model zeolites can range from mean-field diffusion theory to single-file diffusion as the degree of diffusion anisotropy ranges from one extreme to the other. From this model we were able to provide a simple explanation of why mean-field theory works so well for the zeolite Na-Y-benzene host-guest system. We were also able to formulate a new theory for single-file diffusion in zeolites. The single-file problem is extremely challenging computationally because counter-diffusion results from a global diffusion mode that spans the system.
Since then, the technique has gained wide-spread use in the zeolite literature where a wide variety of zeolite problems have been investigated ranging from the basic problem of single component diffusion through multi-component systems, reactive catalytic systems and catalyst deactivation. Recently we have returned to the issue of tracer diffusion in model zeolites (see figure). A key feature of this problem is that Fick's law of diffusion becomes a matrix equation with two eigenmodes, one corresponding to single component transport diffusion (co-diffusion) and the other to self-diffusion (counter-diffusion).
A steady-state method, tracer counter-permeation, (TCP), has been developed for investigating the counter-diffusion eigenmode of diffusion. The advantage of TCP over all other methods is that the counter-diffusion eigenmode (corresponding to self-diffusion) can be investigated directly using Fick's law under steady state conditions. Recently, we have investigated TCP in anisotropic zeolites using kMC. These studies have shown that TCP in model zeolites can range from mean-field diffusion theory to single-file diffusion as the degree of diffusion anisotropy ranges from one extreme to the other. From this model we were able to provide a simple explanation of why mean-field theory works so well for the zeolite Na-Y-benzene host-guest system. We were also able to formulate a new theory for single-file diffusion in zeolites. The single-file problem is extremely challenging computationally because counter-diffusion results from a global diffusion mode that spans the system.
 In my research, simulation techniques have been developed that are capable of modeling the self-assembly of polymeric and surfactant systems, and adsorption, diffusion and reaction in zeolite catalysts. As a result of these simulations, new theories have been presented for polymer conformations in two-dimensional solutions and melts and for single-file diffusion in zeolites. Entropic repulsion has been identified as an ordering force in hydrophilic-core star copolymers in dilute solution.
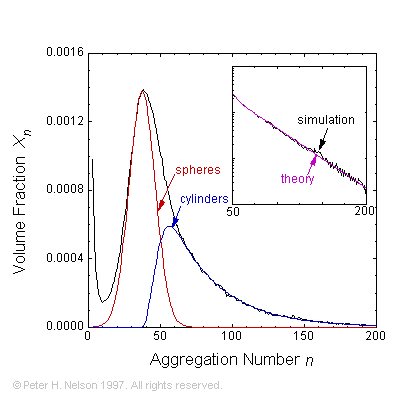
Phenomenological theories and speculations about the micro-structure of self-assembled surfactant and polymer systems have been verified by simulating model systems. We have shown that surfactants spontaneously self-assemble into aggregates (micelles) having a size distribution of the form predicted by rather simple phenomenological theories. The phase behavior of water-surfactant-oil systems has been explained in terms of micellar growth into worm- or thread-like objects.
In my research, simulation techniques have been developed that are capable of modeling the self-assembly of polymeric and surfactant systems, and adsorption, diffusion and reaction in zeolite catalysts. As a result of these simulations, new theories have been presented for polymer conformations in two-dimensional solutions and melts and for single-file diffusion in zeolites. Entropic repulsion has been identified as an ordering force in hydrophilic-core star copolymers in dilute solution.
Phenomenological theories and speculations about the micro-structure of self-assembled surfactant and polymer systems have been verified by simulating model systems. We have shown that surfactants spontaneously self-assemble into aggregates (micelles) having a size distribution of the form predicted by rather simple phenomenological theories. The phase behavior of water-surfactant-oil systems has been explained in terms of micellar growth into worm- or thread-like objects.
In my current research, I am adapting my knowledge of permeation through zeolite membranes to the fascinating area of permeation of small inorganic ions through the ion channel proteins located in nearly all biological membranes. I have recently had something of a breakthrough in this area...
...watch this space for news as it becomes available!
Selected Publications
For more information visit:
 Circle4.com
Circle4.com